Emerging therapies in pancreas cancer
Roswell Park Cancer Institute, Buffalo, NY

|
Review Article
Emerging therapies in pancreas cancer
Roswell Park Cancer Institute, Buffalo, NY
|
|
Abstract
Pancreas cancer has a grave prognosis and treatment options remain limited despite advancement in anti-cancer chemotherapeutics.
This review provides an overview of the emerging therapies for pancreas cancer, focusing on novel signal
transduction inhibitors (insulin-like growth factor receptor, hedgehog/Smo, PI3k/Akt/mTOR) and cytotoxics (nabpaclitaxel)
that are currently in clinical development. Despite the impact molecularly targeted agents have on other
tumor types, their application without cytotoxics in pancreas cancer remains limited. In addition, recent report of the
superiority of an intensive cytotoxic regimen using fluorouracil, irinotecan and oxaliplatin (FOLFIRINOX) over gemcitabine
reminded us of the importance of cytotoxics in this disease. As such, the future of pancreas cancer therapy may be
combination regimens consisting of cytotoxics and molecularly targeted agents.
Key words
Pancreas cancer, chemotherapy, target therapy
J Gastrointest Oncol 2011; 2: 93-103. DOI: 10.3978/j.issn.2078-6891.2011.002
|
|
Introduction
Pancreas cancer is a lethal disease with mortality closely
mirroring the incidence. Approximately 43,410 new cases
will be diagnosed in the United States and 36,800 will die
from the disease in 2010 (1). The mortality rate has not
improved since the 1970s. A number of genetic mutations,
such as KRAS, p16/CDKN2A, TP53, and SMAD4/DPC4,
have been linked to aberrant cell proliferation, signaling,
and reduced apoptosis in the disease (2). Recent genomewide
analysis showed that the genetic makeup of pancreas
cancer is highly complex, with each tumor harboring more
than 60 mutations (3). These aberrancies may be broadly
categorized into 12 core cell-signaling pathways involved
in the initiation and maintenance of malignant phenotype
in pancreas tumors. These inter-related pathways function
as intracellular ‘highways’, transmitting signals between
extracellular events and the nucleus, and are amendable to
therapeutic interventions (4).
Advancement in molecular biology has increased our
understanding of these anomalies and identified a large
number of molecular targets, against which a large number
of anti-cancer agents had been evaluated during clinical
trials. Despite this, erlotinib, a tyrosine kinase inhibitor
(TKI) against epidermal growth factor receptor, is the only
drug after gemcitabine approved by US Food and Drug
Administration for the treatment of advanced pancreas
cancer (5). Approaches to target angiogenesis using agents
such as bevacizumab and sorafenib have failed to achieve
improvement (6-9). Reasons for the failure are likely
multifactorial, including the wrong target, problems in drug
delivery, the existence of resistance or redundant molecular
pathways and failure to identify the susceptible molecular
phenotype. In this review, we will focus primarily on the
classes of targets and corresponding drugs currently in
clinical evaluation that may have potential impact on the
life of pancreas cancer patients in the near future (Table 1).
Agents targeting epidermal growth factor receptor (EGFR)
and vascular endothelial growth factor receptor (VEGFR)
pathways have been reviewed in detail by other authors and
we will discuss them briefly here (Figure 1).
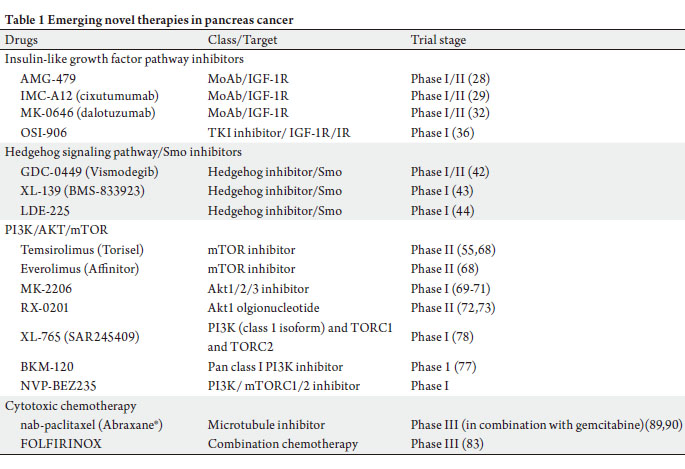 
Figure 1 Signaling pathways implicated in pancreas carcinogenesis. Agents against these pathways are under clinical investigation.
|
|
Human epidermal growth factor pathway
The human epidermal growth factor receptor pathway
family includes EGFR (ErbB-1), HER2/neu (ErbB-2),
HER3 (ErbB-3) and Her4 (ErbB-4). EGFR is an attractive target in pancreas cancer due to its frequency, higher grade
and that increased expression associated with a worse
prognosis (10,11). In a randomized trial of erlotinib plus
gemcitabine versus gemcitabine alone, patients receiving
the combination has a statistically significant improvement
in overall survival (0.82 HR, 6.24 months vs 5.91 months)
(5). However, the improvement is marginal and many
oncologists consider the 2 weeks survival improvement
unsatisfactory. The inhibitor is being evaluated in the
adjuvant setting, and in combination with other targeted
agents such as insulin-like growth factor (IGF) pathway
targeting drugs.
Cetuximab is a monoclonal antibody (MoAb) against
the ligand-binding domain of the EGFR evaluated in
combination with gemcitabine in a randomized phase
III trial. However, the study failed to demonstrate the
superiority of the combination over the gemcitabine
control arm (12). Subset analysis showed that tumor
EGFR e x pres sion does not predic t benef it to the
cetuximab-containing regimen. A phase II trial with
cetuximab +/- gemcitabine and cisplatin showed similar
negative results (13). The objective response rate was
17.5% for the combination arm versus 12.2% in control, and median progression-free and overall survivals were
4.2 months vs 3.4 months, and 7.8 months vs 7.5 months
respectively.
|
|
Anti-angiogenesis
Pancreas cancer was thought to thrive on neovascularization
and dependent on a rich blood supply as the tumors grow
(14). The importance of vascular endothelial growth factor
(VEGF) pathway was shown in preclinical pancreas cancer
studies (15). Though the exact mechanism in patients is
unclear, anti-angiogenic therapies are thought to interrupt
tumor neovascularization and normalize existing inefficient
tumor vasculature, thereby enhancing drug delivery and
synergize the effects of cytotoxic agents.
Bevacizumab, a MoAb to VEGF ligand was studied
in multiple trials. Recently published CALGB 80303
(gemcitabine +/- bevacizumab) treated 535 patients
and overall response rates, median OS and PFS were
13%, 5.8 months, and 3.8 months for the gemcitabine/
bevacizumab arm and 10%, 5.9 months, and 2.9 months
for the gemcitabine/placebo arm, respectively (16).
When bevacizumab was eva luated in combination with gemcitabine and erlotinib, the phase III trial
failed to demonstrate significant improvement by the
bevacizumab-conta ining arm compared to control
(median OS 7.1 months vs 6.2 months respectively) (8).
Bevacizumab failed to improve survival when evaluated in
combination with gemcitabine and capecitabine in a phase
II trial (6). Despite the intial excitement, bevacizumab
failed to improve survival in advanced pancreas cancer
patients when evaluated in combination with standard of
care.
A number of small molecular tyrosine kinase inhibitors
against VEGFR2, including sorafenib, sunitinib and
vatalatinib, have being evaluated in the disease but none
showed positive efficacy signal so far (6-9). Combination
therapies targeting VEGFRs and other signaling pathways are under investigation.
|
|
Insulin-like growth factor pathway
The IGF axis comprises multiple circulating ligands, such
as IGF-1, IGF-II and insulin, interacting with membrane
bound receptors, such as type I IGF receptor (IGF-1R).
The PI3k-Akt pathway is one main downstream mediator
of IGF-1R signaling and plays a potentially important
role in anticancer drug resistance (17). IGF-1R has been
shown in preclinical studies to mediate resistance to EGFR
inhibition, and co-targeting of both receptors enhances
the abrogation of PI3k-Akt activity and reduces survivin
expression (18,19). Transgeneic mouse models of pancreas
cancer expressing high levels of IGF-1R showed increased invasive carcinomas and lymph node metastases (20).
Targeting of IGF-1R expression by siRNAs achieved
growth inhibition in many gastrointestinal malignancies,
suggesting potential importance of the pathway in pancreas
cancer (21-24). In concert, changing IGF-1R copy number
by cDNA plasmid augmented mitogenic response in mouse
embryo. Treatments with MoAb seemed to lead to IGF-1R
internalization and degradation, and enhanced cytotoxic
chemotherapy effects (25). DNA repair pathways are other
downstream effectors of IGF-1R axis and provide the
rationale for combining IGF-1R inhibitors with cytotoxics
(30,31).
A number of agents targeting IGF-1R, both MoAbs and
TKIs, are been evaluated clinically and we are just starting
to understand their clinical role and potential mechanisms
of resistance to this class of drugs (26).
Anti-IGF-1R monoclonal antibodies
AMG-479 is a fully humanized MoAb that blocks the
binding of IGF-I and IGF-II to IGF-1R (IC5027). AMG-479 completely inhibited l igandinduced
dimerization and activation of IGF-1R/IGF-
1R and IGF-1R/IR in two pancreas cancer cell lines. The
antibody reduced IGF-1R-mediated downstream Akt
phosphorylation with pro-apoptotic and anti-proliferative
effects in the cancer cell lines. The agent demonstrated
additive effects with gemcitabine in preclinical studies (27).
In a randomized phase II trial, AMG-479 in combination
with gemcitabine demonstrated a trend to improvement in
median survival when compared to the placebo/gemcitabine
control arm (8.7m vs 5.9m; HR 0.67, P=0.12) in previously
untreated metastatic pancreas cancer patients. The median
PFS was 5.1 months and 2.1 months respectively (HR 0.65,
P=0.07). The investigators conclude that there was sufficient
efficacy signal to warrant further evaluation in a phase III
trial.
IMC-A12 (cixutumumab) (29) and MK-0646
(dalotuzumab) are other anti-IGF-1R MoAb that are being
evaluated in untreated metastatic pancreas cancer patients.
MK-0646 enhanced gemcitabine induced apoptosis in
preclinical studies and is being evaluated clinically. This
phase I/II trial is enrolling patients to 3 treatment arms;
A: gemcitabine 1000mg/m2 weekly × 3 with MK-0646
weekly × 4, Arm B: gemcitabine + MK-0646 + erlotinib
100mg daily, Arm C: gemcitabine 1000mg/m2 weekly ×
3 + erlotinib 100mg daily. MK-0646 achieved 6 partial
responses (PR), 1 hepatic complete response (CR) and 8
stable disease (SD) out of 22 patients (32). Grade 3 or doselimiting
toxicities were rare and included hypergylcemia,
hepatic transaminitis, and febrile neutropenia. The demonstrated responses confirm the hypothesis of crosstalk
between EGFR and IGF a x is signaling and the
importance of adding cytotoxic therapy.
Small molecule IGF-1R/IR kinase inhibitors
Compensatory activation of IR signaling following
inhibition of IGF-1R is emerging as a pathway of resistance
to IGF-1R MoAbs. TKIs against IGF axis thus have a
theoretical advantage over MoAbs given the IR cross
reactivity (33). OSI-906 is a potent and highly selective
inhibitor of IGF-1R, with 14 times greater selectivity for
IGF-1R over IR.34 OSI-906 alone did not show significant
efficacy in pancreas cancer cell lines and was further
evaluated in other tumor types preclinically (35). IGF-
1R pathway has been reported as potential resistance
mechanism to EGFR inhibition and it seems logical to
expect increased efficacy when an IGF-1R inhibitor is
combined with gemcitabine and erolitinib in pancreas
cancer patients. Clinical trials evaluating OSI-906 with
gemcitabine and erlotinib combination have yet to be
initiated. However, the dosing regimen and tox icity
profile of the combination of OSI-906 and erlotinib were
reported at 2010 American Society of Clinical Oncology
Annual Meeting: OSI-906, administered daily at 50mg
and 100mg, combined with erlotinib 100mg daily yielded
stable disease in 4 out of 7 (57%) patients, including
adrenocortical carcinoma, Ewings sarcoma, chordoma
and adenocarcinoma of unknown primary (36). Toxicities
included fatigue (31%) gastrointestinal side effects diarrhea
(31%) nausea (15%); grade ≥3 hyperglycemia.
|
|
Hedgehog/smoothened pathway
Smoothened (Smo) is a transmembrane receptor with seven
domains, and the activity is repressed by Patched (Ptch).
The repression is relieved when ligands bind to Ptch or when
there is activating mutations in Ptch, leading to increased
transcription and up-regulation of Gli-1 to 3, thereby
modulating cell cycle and adhesion, angiogenesis, and
apoptosis. In a comprehensive genomic analysis of pancreas
cancers, mutations in at least one Hedgehog (Hh) signaling
component has been reported in all samples analyzed,
indicating the importance of Hh pathway in pancreas
tumorgenesis (3). In addition, Hh signaling may be an
important modulator of tumor-stromal interaction in the
disease (37,38). Preclinically, Olive et al. evaluated IP-926,
a Smo inhibitor, with gemcitabine which the combination
improved survival of tumor-bearing mice and reduced
metastasis in a transgenic model (39). The anti-cancer
effect seems to be related to a decrease in tumor-associated
stromal tissue and improve drug delivery by stimulating VEGF-independent angiogenesis. In this study, the tumorbearing
mice eventually adapted to chronic Smo inhibition
and became resistant to the treatment, thus raising the
importance in identifying potential resistant mechanisms.
Hh signaling is also implicated as an important mediator
of cancer stem cell (CSC) phenotype in pancreas cancer.
Several groups have reported on the cellular markers of
CSCs in pancreas cancer and the CSCs may be identified
by the co-expression of CS133/CXCR4, or CD44/CD24/
ESA. Extractions enriched in cancer cells expressing
these markers is highly tumorigenic in in vitro and in vivo
experiments and re-capitulate the characteristics of parent
tumors (40,41). Analysis of the CSCs found increased
activation of Hh signaling and other self-renewal signaling
pathways. Mueller et al reported anti-CSC effects when
pancreas tumors were treated with a combination of
cyclopamine or CUR199691 (Smo inhibitors), rapamycin
(mTOR inhibitor) and gemcitabine, and treated tumorbearing
mice survived longer than control (40). This was
associated with elimination of CD133-expressing CSCs.
As such, approaches targeting CSC signaling pathways are
worth exploring clinically.
GDC-0449 (Vismodegib), XL139 (BMS-833923),
and LDE225 are oral agents with anti-Smo activities in
low nanomolar range, and skin Gli-2 expression has been
used a potential pharmacodynamic markers for this class
of agents. Known side effects of Hh inhibitors include
dysguesia, nausea, muscle spasms, rhabdomyolysis, and
alteration in cholesterol biosynthesis. GDC-0449 is furthest
in development and clinical trials evaluating the efficacy
in combination with gemcitabine and nab-paclitaxel or
gemcitabine with and without erlotinib in previously
untreated advanced pancreas cancer patients are starting
soon (42). The clinical efficacy of Smo inhibitors in pancreas
cancer remains unclear from the single-agent phase I trials
conducted so far (43,44). The ability of Hh inhibitors to
reduce stromal tissue and enhances the delivery of cytotoxic
drugs in preclinical studies may be exploited to enhance the
response rate in pancreas cancer patients. Such treatment
has the potential of benef iting patients with locally
advanced or borderline resectable disease (45).
Potential mechanism of resistance to Smo inhibitors can
be learnt from medulloblastoma models, which has been
linked to alteration in the binding site of Smo by GDC-0449
(46). For LDE225, resistance may be related to a number
of factors including Gli2 chromosomal amplification
(a downs t ream ef fec tor of Smo), upreg u l at ion of
compensatory pathways including PI3K/AKT/mTOR, IGF,
and EGFR and, more rarely, point mutations in Smo that
led to reactivated Hh signaling and restored tumor growth
(47). The resistance may be reversed by co-treatment with agents targeting the PI3K/AKT/mTOR, IGF-axis, or EGFR
pathways.
|
|
PI3K/AKT/mTOR pathway
The phosphoinositide 3’-kinase (PI3k)/Akt/mammalian
target of rapamycin (mTOR) pathway acts as a cellular
sensor for nutrients and growth factors, and integrates
signals from multiple receptor kinases to regulate cellular
growth and metabolism (4). The pathway is regulated by
a number of upstream proteins including KRas, which
activating mutations are found in the majority of pancreas
cancer (48). In addition, Akt2 activation, associated
with the development of human cancers, is detected
in about half of the tumors (49). PI3K/Akt/mTOR
activation was associated with early carcinogenesis and
interruption of the pathway achieved anti-proliferation,
-survival, -angiogenic and pro-apoptotic effects (50-58).
Other activating events include PTEN loss and AKT
amplification (59-61). Activation of this pathway was
associated with poor prognosis and contr ibuted to
chemoresistance in many cancers (62-66). Thus, the PI3k/
Akt/mTOR pathway is an attractive pathway to target in
pancreas cancer.
mTOR inhibitors
Everolimus 10mg daily was evaluated in 33 metastatic
gemcitabine-refractory pancreas cancer patients (67).
No objective responses (complete and partial) were
reported and 21% had stable disease at the time of first
surveillance CT scan. Median PFS and OS were 1.8 and
4.5 months respectively. In two smaller clinical trials, 4
gemcitabine-refractory patients received temsirolimus
(CCI-779) and 16 received a combination of everolimus
(30mg once weekly) and erlotinib (150 mg daily) (68).
The former study with temsirolimus was halted due
to toxicities and no objective response was observed,
and the median PFS was 19 days and survival 44 days.
The everolimus and erlotinib combination was better
tolerated, but no response was observed and median PFS
and survival was 49 days and 87 days respectively. These
trials demonstrate that mTOR inhibition as a single agent
is ineffective and combining inhibitors of multiple steps
and the role for these inhibitors may lie in combination
regimens.
Akt inhibitors
Akt inhibitors are another class of agents that abrogate Akt/
mTOR signaling. MK-2206, an allosteric Akt1-3 inhibitor,
was evaluated in a phase I trial of 70 patients with advanced
cancers (69). Interestingly, tumor shrinkage (23%) was obser ved in a patient with PTEN-negative pancreas
cancer and was associated with a 60% decrease in CA19-9.
MK-2206 is being evaluated as weekly (300mg) and every
other day (75mg and 90mg) dosing schedules. MK-2206 is
also being evaluated in combination with cytotoxic chemoagents
and inhibitors of c-Met and EGFR (70,71).
RX-0201 is an antisense oligonucleotide against Akt1
mRNA, thereby interrupting the pathway’s activation. The
anti-sense oligonucleotide demonstrated activity against
pancreas cancer cell lines in low nanomolar range, reducing
the expression of Akt1 mRNA and protein. In in vivo studies,
RX-0201 treatment led to complete response in 2 out of 3
pancreas tumor-bearing mice (72). As such, RX-0201 in
combination with gemcitabine is currently being evaluated
in a phase II trial for metastatic pancreas cancer patients
(73). Given the short half-life typical of anti-sense agents,
RX-0201 is being administered by continuous infusion for
14 days of a 21-day cycle and presents a potential obstacle to
patient accural. Liposomal formulations are in development
(74).
PI3K inhibitors
XL147 and BKM120 are oral class I PI3k inhibitors that are
being evaluated in phase I trials, alone and in combination
therapies (75-77). These trials have focused on lung,
colorectal and breast cancers given the higher frequency of
pathway aberrations in these tumor types. XL765 is a novel
selective inhibitor that interrupts the pathway at various
nodes: PI3K, TORC1 and TORC2. The efficacy of such
agents in pancreas cancer is to be evaluated (78).
|
|
Cytotoxics
Gemcitabine has been the chemotherapy backbone for
the treatment of newly diagnosed advanced pancreas
cancer (79,80). Various other cytotoxic drugs had been
tested in combination with gemcitabine, including
f luoropyrimidines, platinum derivatives, and taxanes
(80-84). Meta-analysis of various cytotoxic trials over the
last one-and-a-half decades suggest improved survival with
doublet or triplet gemcitabine-based therapy among patients
with good performance status, who can, supposedly, better
withstand the toxicities (85).
Final results from the interim analysis of the
PRODIDGE 4/ACCORD 11 trial were presented at 2010
European Society for Medical Oncology annual meeting,
which randomized 342 patients with previously untreated
metastatic pancreas cancer to receiving FOLFIRINOX
(oxaliplatin 85 mg/m2 Day 1 + irinotecan 180 mg/m2 Day
1 + leucovorin 400 mg/m2 Day 1 followed by 5-flurouracil
400 mg/m2 bolus Day 1 and 2,400 mg/m2 46 hours continuous infusion biweekly) or gemcitabine alone. The
study was stopped on recommendation by the independent
monitoring committee during preplanned interim analysis
when FOLFIRINIOX was determined to be superior
to gemcitabine alone, making the f luoropyrimidinebased
regimen first non-gemcitabine based regimen to
show significant improvement in overall survival. The
objective response rate for FOLFIRINOX, compared
to gemcitabine alone, was 31.6% vs 9.4% (P=0.0001),
median PFS 6.4 vs 3.3 months (P<0.0001) and median
survival 11.1 vs 6.8 months (HR=0.57, 95% CI =0.45-0.73;
P<0.001) respectively. However, there were significantly
more grade 3 and above toxicities in the FOLFIRINOX
arm, including diarrhea, nausea, vomiting, neuropathy,
neutropenia, neutropenic fever. Given the higher frequency
of clinically significant toxicities, FOLFIRINOX cannot
be accepted as the standard first-line treatment for all
newly diagnosed advanced pancreas cancer patients. The
choice of FOLFIRINOX in advanced patients needs to
be personalized according to factors such as performance
status, treatment aim, physiological reserve and patient
preference, and the role in adjuvant setting is being
evaluated.
Nab-paclitaxel (Abraxane®; Abraxis) is a nano-particle
preparation in which paclita xel is bound to albumin
as compared to sb-paclitaxel (Taxol®, Bristol Meyers
Squibb), which is dissolved in poloxyethylated castor
oil (Cremaphor EL®) and ethanol. The absence of castor
oil renders nab-paclitaxel clinically advantageous since
this avoids the infusion and hypersensitivity reaction
characteristics of sb-paclitaxel. In the initial phase I clinical
trial of nab-paclitaxel, there was no hypersensitivity
reaction typical of sb-paclitaxel and was well tolerated up
to 300mg/m2 administered as a 30-minute infusion (86).
The recommended dosing for nab-paclitaxel is 260mg/
m2 compared to 175 mg/m2 for sb-paclitaxel (87). In a
crossover pharmacokinetic study to limit patient variability,
nab-pacliataxel had higher peak plasma and unbound
concentrations (88). Greater unbound fraction of paclitaxel
has been hypothesized to lead to greater efficacy seen in
many clinical trials.
One possible mechanism of efficacy by the albuminbound
agent may be related to enhanced tumor uptake
through interaction with the SPARC (secreted protein
acid rich in cysteine) molecule. The SPARC gene, highly
conserved among vertebrates, regulates the assembly,
organization, and turnover of the extracellular matrix
by binding and modulating the deposition of multiple
structural components and attenuating the activity of
extracellular proteases. SPARC is expressed in cancerassociated
stroma and in malignant cells of some types, affecting tumor development, invasion, metastases,
ang iogene si s and inf l ammat ion. SPARC-induced
changes in the tumor microenvironment can suppress
or promote progression of different cancers depending
on the tissue and cell type. SPARC expression is related
to tumor aggressiveness though the exact mechanism is
unclear. The molecule regulates the effects of bFGF and
VEGF on MAPK signaling and increased expression of
SPARC in pancreas tumors has been related to poorer
survival (91,92). Infante et al. characterized SPARC
expression in peritumoral f ibroblasts and pancreas
cells from 299 patients with resectable pancreas cancer.
Median sur vival was halved in patients’ tumors that
expressed SPARC (15 months vs 30 months) and when
cases were controlled for other prognostic factors (tumor
size, positive lymph nodes, margin status, tumor grade,
and age) the hazard ratio (HR) was significant (HR 1.89;
95% CI, 1.31 to 2.74).
Therapies combining nab-paclitaxel with gemcitabine
are under investigation in pancreas cancer given the high
expression of SPARC in pancreas cancer. Several studies
are underway and preliminary result showed impressive
responsive rate and encouraging survival outcome. In a
phase I/II trial, 63 previously untreated metastatic patients
were treated with nab-paclitaxel and gemcitabine and
among the 49 evaluable patients, 1 achieved CR (2%), 12
PRs (24%) and 20 SD (41%) (clinical benefit rate 67%). The
response rate and PFS correlated with SPARC expression
by immunohistochemistr y (89). A single institution
retrospective review of this combination in neoadjuvant
setting for borderline and unresectable patients confirmed
the high response rate (69% PR and 23% SD). About 23%
of patients in the study went on to surgical resection with
curative intent (90). This regimen is being evaluated in a
phase III randomized trial among patients with untreated
metastatic pancreas cancer.
|
|
Conclusion
Despite advancement in anti-cancer therapeutics, treatment
options remain limited and prognosis poor for patients
with pancreas cancer. The molecularly targeted agents held
significant promise in pancreas cancer for several reasons,
including the better-tolerated toxicity profiles and they
target known molecular aberrancies. However, strategies
to target angiogenesis and EGFR pathways had, in general,
not being successful and the underlying reasons remain
unclear. Other exciting molecular targets that can be
interrupted by clinical grade drugs include the IGF, Hh and
PI3k/Akt/mTOR pathways. As these agents complete early phase evaluation, their role in the treatment of pancreas
cancer will be evaluated either alone or in combination
therapies. Importantly, in-depth correlative studies using
patient blood and tumor samples should be incorporated
to better select the patient population most likely to benefit
from these agents and also, to understand the mechanism of
efficacy (or futility).
An important recent development is the demonstration
of the superiority of intense cytotoxic regime n
(FOLFIRINOX) over gemcitabine alone in previously
untreated pancreas cancer patients. Though the regimen
can hardly be accepted as the standard for advanced disease
due to its significant side effect profile, the trial points to
the continual importance of cytotoxic agents in treating
the disease. As such, one eagerly awaits the result from the
phase III trial of nab-paclitaxel plus gemcitabine versus
gemcitabine alone in metastatic pancreas cancer patients
given the encouraging result so far.
|
|
References
Cite this article as:
Kotowski A, Ma W. Emerging therapies in pancreas cancer. J Gastrointest Oncol. 2011;2(2):93-103. DOI:10.3978/j.issn.2078-6891.2011.002
|

