|
Introduction
Modern anti-cancer therapies using specific kinase inhibitors
are directed towards critical molecular targets that are involved
in tumor progression and resistance towards cytotoxic agents.
These therapies have led to modest incremental benefit for
unselected cancer patients over that offered by the traditional
cytotoxic agents. Significant benefit from these novel kinase
inhibitors is limited to a select few patients who may have
activating mutations related to the target kinases. Oncologists
and clinical investigators have long been aware of the interindividual
differences in prognosis and therapeutic outcome
of similar cancer histologies. These differences are attributable
to the genetic and epigenetic heterogeneity of cancer. There
has therefore been a recent emphasis on a more personalized
treatment approach based on the underlying genetic profile
(1). Personalized therapies, wherein underlying genetic or
pathway aberrations are matched with specific therapeutic
agents, are likely to change the existing treatment paradigms
and lead to exponential clinical gains. The opportunities for targeted therapeutics in cancer
at the current time are considerable. However, there are
also a number of challenges in this field. The success of a
personalized approach depends upon the identification
of the underlying molecular abnormality using a reliable
biomarker. Clinical trials of personalized therapy for cancer
using standard randomized trial designs are not inexpensive, and the current regulatory standards for drug approval do
not sufficiently address the personalized therapy paradigm.
Furthermore, there are ethical issues involved in the design
of randomized clinical trials for a specific, targeted patient
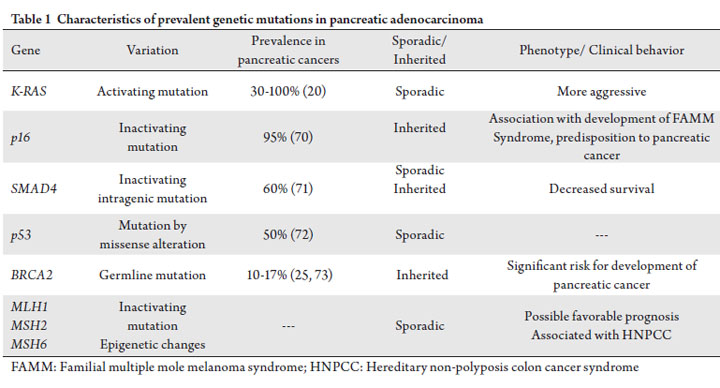
population. Pancreatic cancer is one of the most genetically
heterogeneous of human cancers and may be particularly
suited for personalized therapy.
Success in personalized cancer therapies
Personalized medical care in oncology is currently a reality
for a select group of cancers. With improved knowledge of
tumor biology and the advent of novel technologies allowing
identification of molecular targets, it has become possible to
develop therapies against different subsets of cancers. Specific
examples are discussed below.
• The recognition of biologic and molecular subtypes of
breast cancer that have differential responses to therapeutic
agents has had a major impact in the treatment of this
disease (2). For instance, breast cancers that express
endocrine receptors, in particular the estrogen receptor,
derive benefit from endocrine therapy and may be more
responsive to pre-operative chemotherapy (3-5). About
20-25% of the breast cancers overexpress the human
epidermal growth factor receptor (HER2). Tumor
HER2 status predicts resistance to chemotherapeutic
and hormonal agents and is associated with aggressive
tumor biology (6). Moreover, tumors that express high
levels of HER2 benefit from the HER2-targeted agents,
trastuzumab and lapatinib (7). • The development of imatinib mesylate has revolutionized
the treatment of BCR-ABL-positive chronic myelogenous
leukemia (CML). Targeting the phosphorylation of the BCR-ABL fusion protein, which is critical to CML cell
growth and survival, this highly effective multi-tyrosine
kinase inhibitor is offered as initial therapy to all patients
presenting with CML, inducing durable complete
cytogenetic response in up to 80-85% of patients. Since
its introduction in 2001, imatinib has replaced interferon
based therapies and decreased the need for the highly
morbid procedure of allogeneic stem cell transplantation,
which are now reserved for nonresponders to imatinib
or for those with intolerable side effects (8). Similarly,
the success of imatinib has also been mirrored for
gastrointestinal stromal tumors, in which the median
survival of these patients has been increased from
approximately 20 to 60 months (9). • In non-small cell lung cancer, the tyrosine kinase
inhibitors, erlotinib and gefitinib, have demonstrated
efficacy by blocking the gene encoding epidermal growth
factor receptor (EGFR) and resulted in clinical benefit in
certain subgroups of patients. Each agent has increased
response rates and progression free survival in patients
harboring activating EGFR tyrosine kinase domain
mutations (10,11). Moreover, a recent large phase III
randomized controlled trial demonstrated the superiority
of first-line gefitinib therapy compared to combination
chemotherapy in a clinically selected population consisting
of Asian patients, women with adenocarcinoma and a light
smoking history (12). The success of these examples demonstrates that patient
outcomes can be improved by use of therapies that are
rationally selected against molecular targets. In each example,
knowledge of the molecular profile of the tumor guided the
selection of therapy for the patient.
Pancreatic cancer is heterogeneous
Pancreatic cancer is genetically and biologically
heterogeneous. There is extensive inter-tumor genetic
variability from individual to individual, resulting in
multiple combinations of genetic mutations. For instance,
it has been demonstrated that the pancreatic cancer genome
is highly complex, with an average of 63 somatic alterations
in each cancer, the majority of which are point mutations
(13). Underlying these large numbers of functional genetic
alterations, however, is the deregulation of 12 core
biological regulatory processes or pathways in the majority
of pancreatic tumors (13). This genetic heterogeneity can
be considered in terms of three main molecular events:
mutational oncogenic activation, tumor suppressor gene
inactivation, and inactivation of genome maintenance genes
involved in repair mechanisms (14). Oncogenic k-ras mutations occur in 30% of early precursor lesions and 90% of advanced pancreatic adenocarcinomas and
represent the most frequently encountered genetic variation
in pancreatic cancer (15). Mouse model studies indicate
that k-ras mutations are an initiating step in pathogenesis
of pancreatic oncogenesis (16), and the prevalence of k-ras
mutations increases with increasing dysplasia in precursor
lesions (17). K-ras is a member of the ras family of GTPbinding
proteins that mediate a wide variety of cellular
functions including differentiation, proliferation and survival
(18). Multiple effector pathways and mediators (RAFmitogen-
activated kinase, phosphoinositide-3-kinase, Ral
GDS pathways and NFĸB) are engaged by k-ras activation,
accelerate oncogenesis and represent potential downstream
therapeutic targets (19). At the current time, we have not
successfully targeted the k-ras activating mutations. However,
its downstream effector molecules have been targeted with
success. The majority of pancreatic tumors have inactivation
of the tumor suppressor genes p16, p53 and SMAD4,
leading to loss of function (20). Inherited p16 mutations
have been implicated in the etiology of the Familial
Multiple Mole Melanoma (FAMMM) syndrome, which
carries an increased risk of developing pancreatic cancer.
Alteration of the p53 tumor suppressor gene, by missense
alterations of the DNA-binding domain, occurs in >50%
of pancreatic adenocarcinomas and disrupt regulation of
cellular proliferation and apoptosis in response to DNA
damage (20). Elevated levels of the calcium-binding protein
S100A2, a potent modulator of p53 transcriptional activity
may correlate with the metastatic phenotype of pancreatic
cancer and a poor outcome following pancreatectomy
(21, 22). Approximately 60% of pancreatic cancers have
inactivation of the SMAD4 gene by processes of homozygous
deletion and intragenic mutation, which are important in the
intracellular mediation of the TGF beta intracellular signaling
pathway. SMAD4 gene mutational status has been shown
to significantly correlate with patient outcome, as pancreatic
cancer patients with loss of SMAD4 expression have a greater
propensity to metastasize and a poorer prognosis (23). As the
SMAD4 protein can be detected by immunohistochemical
staining, SMAD4 mutational status may be useful as a
molecular prognostic marker as well as predictor for TGF
beta-directed therapies. Another tumor suppressor gene of interest is BRCA2,
as inherited loss of function mutations of this gene are
thought to be associated with an increased predisposition to
developing pancreatic cancer and promotion of the malignant
progression of pancreatic neoplasms (24). Estimated to
occur in approximately 10% of pancreatic cancers, germline
inactivation of the BRCA2 gene renders the homologous
recombination repair of DNA cross-linking damage deficient and consequently causes genomic instability (25). In vivo,
BRCA2 deficient xenografts demonstrate hypersensitivity to
DNA crosslinking agents including cisplatin (26). Mutations or epigenetic changes of DNA mismatch
repair (MMR) genes such as MLH1, MSH2, MSH3, MSH6,
PMS1, and PMS2, which play a role in the correction of
DNA replication errors also contribute to the pathogenesis
of pancreatic cancer (27). Microsatellite instability (MSI),
resulting from inactivation of a DNA MMR gene, is more
prevalent in a histologically and molecularly distinct subset of
pancreatic carcinomas (28). Consistent with previous reports
that the prognosis of patients with MSI positive tumors was
better than that of patients with MSI negative tumors in
colorectal cancer (29), gastric cancer (30), and cancer of the
papilla of Vater (31), MSI positivity in pancreatic cancer may
also portend a more favorable prognosis (32). Moreover, the
possibility of a germline mutation and presence of hereditary
non-polyposis colorectal cancer syndrome (HNPCC),
or Lynch syndrome, correlates with presence of defective
MMR and increased susceptibility to developing other
gastrointestinal malignancies. MSI-H colorectal cancers
derive benefit from irinotecan therapy; whether this is also
the case with pancreatic cancer remains to be determined
(33). These unique molecular features of pancreatic cancer
have potential utility of being developed into molecular
prognostic indicators of outcome and as therapeutic targets
while establishing an individualized treatment plan for a
patient. These genetic abnormalities and their incidence are
represented in Table 1. At MD Anderson Cancer Center,
we are investigating the role of pharmacogenetics in the individualization of therapies for pancreatic cancer.
Pharmacogenetics
To personalize therapy, it must be recognized that
considerable inter-individual variability in therapeutic
outcome arises at least partly from the underling genetic
profile which can impact on drug pharmacokinetics and
toxicity profile (referred to as pharmacogenetics) (34).
Modern technologies can allow the investigator to interrogate
the pathway impacted by the study agent (candidate gene
approach) or more recently, the whole genome (genome
wide association studies). Implications of pharmacogenetics
are manifold and include a shift away from current paradigm
of offering a standard therapy to all patients with a similar
disease phenotype to an individualized treatment plan that
accounts for pharmacogenetic profile. However, the ethical,
legal, and economic impact resulting from rapid advances
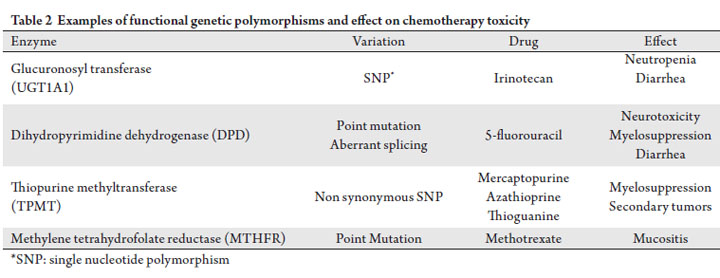
in this field is still yet to be determined. Table 2 depicts
previously described genetic variations of commonly used
anti-cancer agents that are presently available for clinical
management. We have investigated the variations of genes involved in
the metabolism of gemcitabine, the most commonly utilized
agent for pancreatic cancer.
Advanced pancreatic cancer - SNP data – gemcitabine
Despite its role as the backbone of pancreatic cancer therapy,
gemcitabine demonstrates only a modest response at the expense of hematologic toxicity which can result in treatment
delays and dose decreases. Many gemcitabine resistance
mechanisms including altered levels of its activation
enzyme, decreased intracellular drug transport, increased
drug metabolism, and increased expression of DNA repair
enzymes have been proposed as contributing to the failure
of gemcitabine therapy (35-38). Evidence published in early
2009 from the RTOG9704 trial confirmed that increased
intra-tumoral expression of human equilibrative nucleoside
transporter (hENT1), the major protein believed to be
responsible for transporting gemcitabine into cells, was
associated with an improved overall and disease-free survival
in patients with resected pancreatic cancer treated with
gemcitabine as compared with those receiving 5-fluorouracil
(39). Preclinical evaluation in lung cancer has demonstrated
that overexpression of ribonucleotide reductase regulatory
subunit M1 (RRM1), a DNA repair enzyme, may also
be a marker of poor response to gemcitabine therapy
(40). Previous clinical studies have suggested that
gemcitabine therapy has less efficacy in patients with
advanced tumors expressing high levels of RRM1 (41, 42).
Further immunohistochemical study of RRM1 correlates
overexpression of protein levels with a worse overall survival
and disease control than those patients with RRM1-negative
tumors (43). Recently, the clinical significance of single nucleotide
polymorphisms (SNP) of gemcitabine metabolic genes was
evaluated in pancreatic cancer by our group (44). Okazaki
et al examined 17 SNPs of eight genes in 154 patients with
potentially resectable pancreatic adenocarcinoma treated
with neoadjuvant concurrent gemcitabine and radiation
therapy with or without cisplatin. Though none of the
SNPs was significantly associated with overall survival (OS)
individually, a combined genotype effect was observed, in which
the risk of death was increased for patients with variant gemcitabine metabolic genes. Moreover, hematologic toxicity
due to gemcitabine was associated with polymorphisms of
the cytidine deaminase and deoxycytidine kinase genes. This
study suggests that the clinical outcome of pancreatic cancer
patients treated with gemcitabine-based chemotherapy
results, in part, from variations in genes responsible for
gemcitabine metabolism and elimination. The results of this
study support the investigation of pharmacogenetic profiling
to individualize gemcitabine-based therapy for pancreatic
cancer. An effort is being made to expand pharmacogenetic
profiling for other agents that are considered effective in
pancreatic cancer. Although gemcitabine has been the mainstay of
chemotherapy for pancreatic cancer for the past decade,
the beneficial effects from gemcitabine are mostly
palliative and survival gains from this agent are limited.
Developmental efforts have focused on the addition of other
chemotherapeutic agents to gemcitabine and thus far the
only phase III study that resulted in survival benefit was the
National Cancer Institute of Canada-Clinical Trials Group
PA.3 (NCIC-CTG PA.3) study which showed that the
addition of erlotinib to gemcitabine resulted in a modestly
improved survival as compared with gemcitabine alone
(45). A recent phase III study presented at the American
Society of Clinical Oncology (ASCO) meeting in 2010,
investigated the combination of 5-fluorouracil, oxaliplatin and
irinotecan (FOLFIRINOX) vs. gemcitabine for the treatment
of patients with advanced pancreatic cancer (46). In this
study, 342 patients were enrolled; at a preplanned interim
analysis, the median overall survival in the FOLFIRINOX
arm was significantly longer than that in the gemcitabine
arm (10.5 vs. 6.9 months, p< 0.0001) at the cost of higher
toxicities including diarrhea, emesis and neutropenia in the
study arm. While the toxicities associated with this regimen
are concerning, there is now an alternative to gemcitabine chemotherapy for pancreatic cancer patients. As discussed
below, there are promising biomarkers that correlate
with gemcitabine resistance and the availability of a valid
alternative regimen that excludes gemcitabine opens avenues
for biomarker-driven cytotoxic chemotherapy in pancreatic
cancer.
Limitations of tissue acquisition in pancreatic cancer
An important limitation in case of biomarkers to study
pancreatic cancer is that tissue procurement is limited in
this disease. A dense fibrotic stroma surrounds the tumor
and most biopsies are obtained via fine needle aspiration.
These aspirates are paucicellular and this limits biomarker
assessment. On the other hand, core needle biopsies are
feasible from metastatic sites such as liver and often yield
adequate tissue for biomarkers. This, however, limits the stage
of cancers for study and introduces a selection bias. Better
technologies to examine biomarkers in the peripheral blood
or from fine needle aspirates are required..
Cancer biomarkers: better indicators of ‘non-responsiveness’
Despite advances in biomarker technology, the currently
available biomarkers are more effective in identifying
patients who will not respond to targeted agents rather than
identify those who will benefit. For instance k-ras mutation
or HER2 neu status of the tumors have thus far been more
effective as a negative predictive markers for cetuximab or
herceptin therapy for colorectal and breast cancers than as
predictors of response. For instance, the response rate for
patients treated with panitumumab in the phase III trial of
panitumumab versus supportive care (BSC) was 10%, but
the retrospective analysis of patients with wild-type k-ras
tumors from that trial demonstrated a response rate to
panitumumab of 17% (47, 48). These results are comparable
with those from the phase III trial of cetuximab versus
BSC, with response rates of 8% for those patients receiving
cetuximab and 12.8% for patients with wild-type k-ras
tumors receiving cetuximab (49). Thus, the response rates
in the k-ras wild type tumors is very modest and the positive
predictive value of this mutation is low; on the other hand
the negative predictive value is higher with no responses
in the mutated phenotype. Cross-talk between signaling
pathways and tumor genetic heterogeneity may account for
these results; tumors that have drug-sensitizing mutations
may have simultaneous activation of down-stream drugresistance
pathways or mutations. Despite these limitations,
biomarker-driven clinical trials are likely to be associated
with stronger efficacy signals and lead to cost-effective
health care. Specific examples as applicable to pancreatic cancer are discussed below.
Biomarkers for erlotinib
Several promising biomarkers of therapeutic interest have
been described in patients with non-small cell lung cancer
treated with gefitinib or erlotinib. These include activating
mutations of EGFR and tumor k-ras mutation status. These
biomarkers have yet to be prospectively validated in the case
of pancreatic cancer. Although the NCIC-CTG PA.3 study
did perform a post-hoc analysis of available pancreatic tumor
biopsy tissue for k-ras mutations and EGFR amplification,
it failed to establish a significant link between either of
these markers and outcome with a trend favoring erlotinib
observed only in patients with the wild-type k-ras (50, 51).
Epithelial to mesenchymal transformation (EMT) has also
been correlated with the efficacy of erlotinib therapy in lung
cancer (better response noted with the epithelial phenotype)
and is a common feature of pancreatic cancers as well. The
degree of EMT is measured by the relative levels of molecular
epithelial (vimetin, integrin-alpha 5) versus mesenchymal
(desmoplakin, keratin-19, cadherin 1) markers (52). The
mesenchymal phenotype, morphologically distinguished by
the irregularity of its cells, lack of organized structure and
weak intracellular adhesions is more aggressive and carries a
poor prognosis (53). Further investigation of the predictive
value of k-ras mutation status and EMT in pancreatic
cancer is needed. Recent data from Ratain et al, indicate
the association between polymorphisms of the multidrug
ABCG2 transporter and erlotinib pharmacokinetic profile
and EGFR polymorphisms and diarrhea (54). Incorporation
of these biomarkers can help reduce the toxicity resulting
from erlotinib therapy.
Nanoparticle albumin-bound (Nab) paclitaxel
Nab-paclitaxel is a solvent-free, albumin-bound 130-nm
particle form of paclitaxel (Abraxane, Abraxis Bioscience, CA,
USA), which was developed to avoid toxicities associated with
the Cremophor vehicle used in solvent-based paclitaxel. This
agent takes advantage of the increased delivery of albumin
to tumors through receptor-mediated transport. SPARC
(secreted protein, acidic and rich in cysteine) is selectively
secreted by pancreatic cancer cells and binds to albuminbound
paclitaxel with the resultant release of paclitaxel in the
vicinity of tumor cells. Together, the absence of solvents and
the receptor-mediated delivery result in decreased toxicity
and increased antitumor activity of nab-paclitaxel compared
with solvent-based paclitaxel. A phase I study of gemcitabine
and nab-paclitaxel has demonstrated impressive response
rates and progression-free survival; in this study responses and progression-free survival correlated with SPARC
expression (55). In the future, the investigational plans are to
administer this agent only for the tumors that have SPARC
expression.
Targeting DNA repair to exploit synthetic lethality
Another potential strategy toward development of effective novel therapy for pancreatic cancer is exploiting the concept of synthetic lethality, a genetic interaction in which the combination of mutations in two or more genes leads to cell death. Cells typically have the ability to repair therapyinduced single strand (SS) and double strand (DS) DNA breaks by the conserved mechanisms of base excision repair (BER) and homologous recombination (HR) repair, respectively (56). Since 10% of patients with pancreatic cancer harbor have germline inactivation of the BRCA2 gene, leading to deficient HR, these individuals are susceptible to genomic instability after incurring a second insult to BER (23). Moreover, sporadic pancreatic cancer harbor similar repair pathway defects resulting from other genetic mutations or DNA repair and damage response pathways and share this susceptibility “profile of BRCAness” (57). Defective DNA damage and repair pathways are targets
for inhibition of poly (ADP-ribose) polymerase I (PARP-1),
a critical enzyme of DNA repair. PARP-1 is required for the
BER of chemotherapy and radiation-induced DNA single
strand breaks (58). When PARP-1 is inhibited in the presence
of defective HR repair (as in BRCA2 mutations or in cancers
exhibiting properties of “BRCAness”), the resultant DNA
damage can be lethal (synthetic lethality) (56, 58). Thus,
PARP inhibition might be a useful therapeutic strategy in the
treatment of certain pancreatic cancers and is currently under
investigation. However, the identification of aberrant DNA
repair in cancer tissue is far from ideal at this point. Promising
leads have been published recently to identify aberrant
homologous recombination in body fluids such as ascites;
these need to be validated in pancreatic cancer (59).
IgF1R as a target in pancreatic cancer
Genetic variations in the insulin-like growth factor (IGF)-axis
may also play a role in the development and progression of
pancreatic cancer. It has been previously demonstrated that
the protein products of these pathway genes (IGF1 receptor,
IGF2 receptor, IGF binding protein family, and insulinreceptor
substrate family) are involved in maintenance and
regulation of tissue homeostasis and regulation of growth,
differentiation and migration (60, 61). In a meta-analyses of
96 studies, circulating levels of IGF1, IGFBP3 (IGF binding
protein), and IGFBP3 A-202C genotype were shown to be important in carcinogenesis and potentially serve as
biomarkers for cancer growth in various human malignancies
through genotype-phenotype correlation analyses (62). In pancreatic cancer, IGF1 may function as a growth factor
(63). IGF1 is upregulated in human pancreatic cancer tissue,
with serum levels elevated in pancreatic cancer patients
(64, 65). We recently noted that genetic variations in the IGF
axis pathway are prognostic in advanced pancreatic cancer
(66). After genotyping 41 SNPs from 10 IGF-axis genes in
over 700 advanced pancreatic cancer patients, we noted that
SNP of the IGF1R, IGF2R, and IRS1 gene were significantly
associated with survival. In a current study that includes
an IGF1R-directed antibody, MK-0646 we have noted a
correlation between IGF1/IGFBP3 ratio and response. These
findings will be confirmed in a wider cohort of patients and a
prospective, biomarker-driven study is planned (67).
Biomarker validation
Biomarker-driven therapeutic clinical trials can include
the co-development of the biomarker and the study agent,
particularly when the biomarker is relatively novel.
The goal is to have appropriate validation before the
marker can reach clinical applications; but validation is a
cumbersome process for which standards are not clearly
established. Critical issues that need to be addressed for the
validation studies include the specificity and reproducibility
of the marker. In the case of pancreatic cancer, this is further
complicated by inter-patient heterogeneity and difficulty in
obtaining representative sampling from the primary tumor
site (pancreas). Regulatory guidance in this regard will be
imperative in the development of biomarker driven targeted
therapies for pancreatic cancer.
Clinical trial design for targeted agents
The use of a panel of biomarkers as potential predictive tools
for the enrollment of patients on clinical trials with targeted
agents requires innovative clinical trial design beyond the
traditional simple randomization. These traditional trial
designs are based on the ‘frequentist’ principles. Frequentist
trial designs are based on the probability of observing
results as being disparate from the expected or the ‘null
hypothesis’. In these frequentist designs, a p value is defined
as the probability that the observed results are sufficiently
disparate from the controls and a p value of < 0.05 is generally
considered as significant. The advantage of the traditional
randomized trials is that these are relatively easy to implement
and they are scientifically robust and focused. However, the
latter is also a potential disadvantage as these trial designs
are inflexible, limiting innovation or modification as the trial proceeds. Furthermore, traditional randomized trials tend to
be large and expensive wherein some patients are needlessly
exposed to inferior therapies. Recent examples of these types
of trials which are with erlotinib for advanced pancreatic
cancer and herceptin in gastric cancer which enrolled>500
patients each and proved that these targeted agents were
of some benefit. On the other hand, a sample size of 600
patients was also required to prove that bevacizumab was
ineffective in pancreatic cancer despite the use of stopping
rules in the trial.
In Bayesian designs, uncertainty is measured as a
probability. Unknown parameters are given a probability
distribution while what is known is taken as a given. However,
once the results of the study become more evident, these
are no longer probabilities and are taken as a given. Thus
these trial designs are inherently adaptive and allow the
investigator to modify trials mid course based on current
data. Thus, Bayesian adaptive designs allow for changes to the
clinical trial based on ongoing progress and allow enrichment
based on the results. These designs are especially suitable
for the development of biomarker-directed targeted therapy.
For instance, the prior distribution of a biomarker profile
may not be known with a great deal of certainty; this can
therefore be hypothesized and refined as the trial develops.
A pharmaceutical company can tie in the decision rules
within the Bayesian trial design to determine the pathway for
drug development. Bayesian designs are extensively being
utilized at MD Anderson Cancer Center, wherein over a
hundred clinical trials are ongoing using these principles. A
detailed review of this trial design is described elsewhere.
The disadvantages of this design is that it is computationally
intensive, restricted to a limited number of centers with
expertise and is not yet widely recognized by regulatory
agencies as an efficient and economical pathway towards
drug development. While these issues appear to be complex,
successful implementation is possible and requires a multidisciplinary
effort. One such an example is an ongoing study
in non-small cell lung cancer at our institution.
Battle trial for non small-cell lung cancer
The recently concluded BATTLE 1 (Biomarker-integrated
Approaches of Targeted Therapy for Lung Cancer
Elimination) phase II clinical trial conducted at MD
Anderson Cancer Center illustrates the potential of Bayesian
adaptive randomization as a study design for evaluating novel
targeted therapies in cancer using personalized biomarker
profiles to guide treatment allocations (Fig 1). First, 97
patients with stage IV non small-cell lung cancer who had
received at least one prior chemotherapy were each assigned
to receive one of four possible drugs (Erlotinib, Vandetanib, Erlotinib + Bexarotene, or Sorafenib) by traditional simple
randomization. Core biopsies of the lung were obtained from
this initial subset of patients and profiled for four biomarkers
(EGFR, KRAS/BRAF, VEGFR-2 and RXR/Cyclin D1).
The primary study endpoint was progression-free survival
at 8 weeks. Interim analysis was conducted to determine the
specific biomarker profiles that predicted a favorable clinical
response in each of the four study arms. These interim results
were used to ‘adapt’ the randomization for the next 158
patients who entered the study. That is, each patient in this
latter subset was assigned to that treatment likely to be most
effective given the biomarker characteristics of the patient’
s tumor. Preliminary results indicate increased survival for
the patients treated in this trial as compared with historical
controls from the same institution who received ‘unselected’
therapy. The National Cancer Institute has recently
underscored the value of bringing innovative methodologies
to the design of biomarker-driven pancreatic cancer clinical
trials, and the focus on personalizing management through
the integration of biomarker correlates prospectively into
BATTLE 1 is one such groundbreaking paradigm that can
certainly be applied to pancreatic cancer (68).
|
|
References
- Potti A, Schilsky RL, Nevins JR. Refocusing the war on cancer: the critical
role of personalized treatment. Sci Transl Med 2010;2:28cm13.[LinkOut]
- Peppercorn J, Perou C, Carey L. Molecular subtypes in breast
cancer evaluation and management: divide and conquer. Cancer
Investigation2008;26:1-10.[LinkOut]
- Guarneri V, Broglio K, Kau SW, Cristofanilli M, Buzdar AU, Valero V,
et al. Prognostic value of pathologic complete response after primary
chemotherapy in relation to hormone receptor status and other factors. J
Clin Oncol 2006;24:1037-44.[LinkOut]
- Berry DA, Cirrincione C, Henderson IC, Citron ML, Budman DR,
Goldstein LJ, et al. Estrogen receptor status and outcomes of modern
chemotherapy for patients with node-positive breast cancer. J Am Med
Assoc 2006;295:1658-67.[LinkOut]
- Rouzier R, Perou CM, Symmans WF, Ibrahim N, Cristofanilli M,
Anderson K, et al. Breast cancer molecular subtypes respond differently to
preoperative chemotherapy. Clin Cancer Res 2005;11:5678-85.[LinkOut]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL.
Human breast cancer: correlation of relapse and survival with amplification
of the HER2-/neu oncogen. Science 1987;235:177-82.[LinkOut]
- Hudis CA. Trastuzumab - mechanism of action and use in clinical practice. N Engl J Med 2007;357:39-51.[LinkOut]
- Schiffer C. BCR-ABL tyrosine kinase inhibitors for chronic myelogenous
leukemia. N Engl J Med 2007;357:258-65.[LinkOut]
- Dematteo RP, Ballman KV, Antonescu CR, Maki RG, Pisters PW, Demetri
GD, et al. Adjuvant imatinib mesylate after resection of localised, primary
gastrointestinal stromal tumour: a randomised, double-blind, placebocontrolled
trial. Lancet 2009;373:1097-104.[LinkOut]
- Rosell R, Moran T, Queralt C, Porta R, Cardenal F, Camps C, et al.
Screening for epidermal growth factor receptor mutations in lung cancer. N
Engl J Med 2009;361: 958-67.[LinkOut]
- Mok TS, Wu Y, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib
or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med
2009;361:947-57.[LinkOut]
- Kim ES, Hirsh V, Mok T, Socinski MA, Gervais R, Wu YL, et al: Gefiinib
versus docetaxel in previously treated non-small-cell-lung cancer
(INTEREST): a randomised phase III trial. Lancet 2009;372:1809-18.[LinkOut]
- Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core
signaling pathways in human pancreatic cnacers revealed by global genomic
analyses. Science 2008;321:1801-6.[LinkOut]
- Bert Vogelstein, Kenneth W. Kinzler. The genetic basis of human cancer:
Pancreatic cancer. 2nd ed. New York: McGraw-Hill; 2002.
- Klimstra DS, Longnecker DS. K-ras mutations in pancreatic ductal
proliferative lesions. Am J Pathol 1994;145:1547-50.[LinkOut]
- Moskaluk CA, Hruban RH, Kern SE. p16 and K-ras gene mutations in the
intraductal precursors of human pancreatic adenocarcinoma. Cancer Res
1997;57:2140-3.[LinkOut]
- Jimenez RE, Warshaw AL, Z’graggen K, Hartwig W, Taylor DZ,
Compton CC, et al: Sequential accumulation of K-ras mutations and p53
overexpression in the progression of pancreatic mucinous cystic neoplasms
to malignancy. Ann Surg 1999;230:501-9.[LinkOut]
- Malumbres M, Marbacid M. RAS oncogenes: The first 30 years. Nat Rev
Cancer 2003;3:459-65.[LinkOut]
- Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing
complexity of Ras signaling. . Oncogene 1998;17:1395-413.[LinkOut]
- Rozenblum E, Schutte M, Goggins M, Hahn SA, Panzer S, Zahurak M,
et al. Tumor-suppressive pathways in pancreatic carcinoma. Cancer Res
1997;57:1731-4.[LinkOut]
- Mueller A, Schafer BW, Ferrari S, Weibel M, Makek M, Hochli M, et al.
The calcium-binding protein S100A2 interacts with p53 and modulates its
transcriptional activity. J Biol Chem 2005;280:29186-93.[LinkOut]
- Biankin AV, Kench JG, Colvin EK, Segara D, Scarlett CJ, Nguyen NQ, et
al. Expression of S100A2 calcium-binding protein predicts response to
pancreatectomy for pancreatic cancer. Gastroenterology 2009;137:558-68,
568.e1-11.[LinkOut]
- Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang
X, et al. SMAD4 gene mutations are associated with poor prognosis in
pancreatic cancer. Clin Cancer Res 2009;15:4674-9.[LinkOut]
- Hezel AF, Kimmelman AC, Stanger BZ, Bardeesy N, Depinho RA.
Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev
2006;20:1218-49.[LinkOut]
- Goggins M, Schutte M, Lu J, Moskaluk CA, Weinstein CL, Petersen GM, et al. Germline BRCA2 gene mutations in patients with apparently sporadic
pancreatic carcinomas. Cancer Res 1996; 56:5360-4.[LinkOut]
- van der Heijden MS, Brody JR, Dezentje DA, Gallmeier E, Cunningham
SC, Swartz MJ, et al. In vivo therapeutic responses contingent on Fanconi
anemia/BRCA2 status of the tumor. Clin Cancer Res 2005;11:7508-15.[LinkOut]
- Modrich P, Lahue R. Mismatch repair in replication fidelity, genetic
recombination, and cancer biology. Annu Rev Biochem 1996;65:101-33.[LinkOut]
- Wilentz RE, Goggins M, Redston M, Marcus VA, Adsay NV, Sohn TA,
et al. Genetic, immunohistochemical, and clinical features of medullary
carcinoma of the pancreas: a newly described adn characterized entify. Am
J Pathol 2000;156:1641-51.[LinkOut]
- Gryfe R, Kim H, Hsieh ET, Aronson MD, Holowaty EJ, Bull SB, et al.
Tumor microsatellite instability and clinical outcome in young patients
with colorectal cancer. N Engl J Med 2000;342:69-77.[LinkOut]
- Chiaravalli AM, Cornaggia M, Furlan D, Capella C, Fiocca R, Tagliabue
G, et al. The role of histological investigation in prognostic evaluation of
advanced gastric cancer. Analysis of histological structure and molecular
changes compared with invasive pattern and stage. Virchows Arch
2001;439:158-69.[LinkOut]
- Achille A, Biasi MO, Zamboni G. Cancers of the papilla of vater:
mutator phenotype is associated with good prognosis. Clin cancer Res
1997;3:1841-7.[LinkOut]
- Nakata B, Wang YQ, Yashiro M. Prognostic value of microsatellite
instability in resectable pancreatic cancer. Clin Cancer Res 2002;8:2536-40.[LinkOut]
- Fallik D, Borrini F, Boige V, Viguier J, Jacob S, Miquel C, et al. Microsatellite
instability is a predictive factor of the tumor response to irinotecan in
patients with advanced colorectal cancer. Cancer Res 2003;63:5738-44.[LinkOut]
- Kalow W, Tang BK, Endrenyi I. Hypothesis: comparisons of inter- and
intra-individual variations can substitute for twin studies in drug research.
Pharmacogenetics 1998;8:283-9.[LinkOut]
- Kroep JR, Loves WJ, van der Wilt CL, Alvarez E, Talianidis I, Boven E, et
al. Pretreatment deoxycytidine kinase levels predict in vivo gemcitabine
sensitivity. Mole Cancer Ther 2002;1:371-6.[LinkOut]
- Galmarini CM, Clarke ML, Jordheim Lea. Resistance to gemcitabine in
a human follicular lymphoma cell line is due to partial deletion of the
deoxycytidine kinase gene. BMC Pharmacol 2004;3:8.[LinkOut]
- Achiwa H, Oguri T, Sato S, Maeda H, Niimi T, Ueda R. Determinations of
sensitivity and resistance to gemcitabine: The roles of human equilibrative
nucleoside transporter 1 and deoxycytidine kinase in non-small cell lung
cancer. Cancer Sci 2004;95:753-7.[LinkOut]
- Spratlin J, Sangha R, Glubrecht D, Dabbagh L, Young JD, Dumontet
C, et al. The absence of human equilibrative nucleoside transporter 1 is
associated with reduced survival in patients with gemcitabine-treated
pancreas adenocarcinoma. Clin Cancer Res 2004;10:6956-61.[LinkOut]
- Farrell JJ, Elsaleh H, Garcia M, Lai R, Ammar A, Regine WF, et al.
Human equilibrative nucleoside transporter 1 levels predict response
to gemcitabine in patients with pancreatic cancer. Gastroenterology
2009;136:187-95.[LinkOut]
- Bepler G, Sharma S, Cantor A, Gautam A, Haura E, Simon G, et al. RRM1
and PTEN as prognostic parameters for overall and disease-free survival in
patients with non-small-cell lung cancer. J Clin Oncol 2006;24:4741-7.[LinkOut]
- Ceppi P, Volante M, Novello S, Rapa I, Danenberg KD, Danenberg PV, et al.
ERCC1 and RRM1 gene expression but not EGFR are predictive of shorter
survival in advanced non-small cell lung cancer treated with cisplatin and
gemcitabine. Ann Oncol 2006;17:1818-25.[LinkOut]
- Souglakos J, Boukovinas I, Taron M, Mendez P, Mavroudis D, Tripaki M,
et al. Ribonucleotide reductase subunits M1 and M2 mRNA expression
levels and clinical outcome of lung adenocarcinoma patients treated with
docetael/gemcitabine. Br J Cancer 2008;98:1710-5.[LinkOut]
- Lee JJ, Maeng CH, Baek SK, Kim GY, Yoo JH, Choi CW, et al. The
immunohistochemical overexpression of ribonucleotide reductase
regulatory subunit M1 (RRM1) protein is a predictor of shorter survival to
gemcitabine-based chemotherapy in advanced non-small cell lung cancer
(NSCLC). Lung Cancer 2010 Mar 9. [Epub ahead of print][LinkOut]
- Okazaki T, Javle M, Tanaka M, Abbruzzese JL, Li D. Single nucleotide
polymorphisms of gemcitabine metabolic genes and pancreatic cancer
survival and drug toxicity. Clin Cancer Res 2010;16:320-9.[LinkOut]
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al.
Erlotinib plus gemcitabine compared with gemcitabine alone in patients
with advanced pancreatic cancer: a phase III trial of the National Cancer
Institute of Canada Clinical Trials Group. J Clin Oncol 2007;25:1960-6.[LinkOut]
- Conroy T, Desseigne F, Ychou M, Ducreux O, Bouche R, Guimbaud Y, et al.
Randomized phase III trial comparing FOLFIRINOX (F: 5FU/leucovorin
[LV], irinotecan [I], and oxaliplatin [O]) versus gemcitabine (G) as
first-line treatment for metastatic pancreatic adenocarcinoma (MPA):
Preplanned interim analysis results of the PRODIGE 4/ACCORD 11 trial.
J Clin Oncol 2010;28:15s (suppl; abstr 4010).
- Elez E, Alsina M, Tabernero J. Panitumumab - an effective long-term
treatment for patients with metastatic colorectal cancer and wild-type
KRAS status. Cancer Treat Rev 2010;36:S15-6.[LinkOut]
- Bardelli A, Siena S. Molecular mechanisms of resistance to cetuximab and
panitumumab in colorectal cancer. J Clin Oncol 2010;28:1254-61.[LinkOut]
- Allegra CJ, Jessup JM, Somerfield MR, Hamilton SR, Hammond EH,
Hayes DF, et al. American Society of Clinical Oncology provisional clinical
opinion: testing for KRAS gene mutations in patients with metastatic
colorectal carcinoma to predict response to anti-epidermal growth factor
receptor monoclonal antibody therapy. J Clin Oncol 2009;27:2091-6.[LinkOut]
- Moore MJ, Goldstein D, Hamm J, Figer A, Hecht JR, Gallinger S, et al.
Erlotinib plus gemcitabine compared with gemcitabine alone in patients
with advanced pancreatic cancer: A phase III trial of the national cancer
institute of canada clinical trials group. J Clin Oncol 2007;25:1960-6.[LinkOut]
- Moore MJ, da Cunha Santos G, Kamel-Reid S, Chin K, Tu D, Parulekar W,
et al. The relationship of K-ras mutations and EGFR gene copy number to
outcome in patients treated with erlotinib in the National Cancer Institute
of Canada Clinical Trials Group trial study PA.3. Journal of Clinical
Oncology 2007;25:s4521.
- Kong B, Michalski CW, Hong X, Valkovskaya N, Rieder S, Abiatari I, et al.
AZGP1 is a tumor suppressor in pancreatic cancer inducing mesenchymalto-
epithelial transdifferentiation by inhibiting TGF-beta-mediated ERK
signaling. Oncogene. 2010 Jun 28. [Epub ahead of print][LinkOut]
- Javle MM, Gibbs JF, Iwata KK, Pak Y, Rutledge P, Yu J, et al. Epithelialmesenchymal
transition (EMT) and activated extracellular signal-regulated kinase (p-Erk) in surgically resected pancreatic cancer. Ann Surg Oncol
2007;14:3527-33.[LinkOut]
- Rudin CM, Liu W, Desai A, Karrison T, Jiang X, Janisch L, et al.
Pharmacogenomic and pharmacokinetic determinants of erlotinib toxicity.
J Clin Oncol 2008;26:1119-27.[LinkOut]
- Von Hoff DD, Ramanathan R, Borad M, Laheru D, Smith L, Wood T, et al:
SPARC correlation with response to gemcitabine (G) plus nab-paclitaxel
(nab-P) in patients with advanced metastatic pancreatic cancer: A phase
I/II study. J Clin Oncol 2009; 27: suppl; abstr 4525.
- McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al.
Deficiency in the repair of DNA damage by homologous recombination
and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res
2006;66:8109-15.[LinkOut]
- Turner N, Tutt A, Ashworth A. Hallmarks of “BRCAness” in sporadic
cancers. Nat Rev Cancer 2004;4:1-6.[LinkOut]
- Lord CJ, Ashworth A. Targeted therapy for cancer using PARP inhibitors.
Curr Opin Pharmacol 2008;26:3785-90.[LinkOut]
- Mukhopadhyay A, Elattar A, Cerbinskaite A, Wilkinson SJ, Drew Y, Kyle
S, et al. Development of a functional assay for homologous recombination
status in primary cultures of epithelial ovarian tumor and correlation with
sensitivity to poly(ADP-ribose) polymerase inhibitors. Clin Cancer Res
2010;16:2344-51.[LinkOut]
- Withers DJ, Burks DJ, Towery HHea. Irs-2 coordinates Igf-1 receptormediated
beta-cell development and peripheral insulin signaling. Nat Genet
1999;23:32-40.[LinkOut]
- Dong X, Park S, Lin X, Copps K, Yi X, White MF. Irs1 and Irs2 signaling is
essential for hepatic glucose homeostasis and systemic growth. J Clin Invest
2006;116:101-14.[LinkOut]
- Chen W, Wang S, Tian T, Bai J, Hu Z, Xu Y, et al. Phenotypes and
genotypes of insulin-like growth actor 1, IGF-binding protein-3 and cancer
risk: evidence from 96 studies. Eur J Hum Genet 2009;17:1168-75.[LinkOut]
- Nair PN, De Armond DT, Adamo ML, Strodel WE, Freeman JW. Aberrant
expression and activation of insulin-like growth factor-1 receptor (IGF-1R)
are mediated by an induction of IGF-1R promoter activity and stabilization
of IGF-1R mRNA and contributes to growth factor independence and
increased survival of the pancreatic cancer cell line MIA PaCa-2. Oncogene 2001;20:8203-14.[LinkOut]
- Bergmann U, Funatomi H, Yokoyama M, Beger HG, Korc M. Insulin-like
growth factor I overexpression in human pancreatic cancer: evidence for
autocrine and paracrine roles. Cancer Res 1995;55:2007-11.[LinkOut]
- Karna E, Surazynski A, Orłowski K, Łaszkiewicz J, Puchalski Z, Nawrat
P, et al. Serum and tissue level of insulin-like growth factor-I (IGF-I) and
IGF-I binding proteins as an index of pancreatitis and pancreatic cancer. Int
J Exp Pathol 2002;83:239-45.[LinkOut]
- Dong X, Javle M, Hess KR, Shroff R, Abbruzzese JL, Li D. Insulin-like
growth factor axis gene polymorphisms and clinical outcomes in pancreatic
cancer. Gastroenterology 2010;139:464-73.[LinkOut]
- Javle MM, Varadhachary GR, Shroff RT, Bhosale P, Overman MJ, Weatherly
J, et al. Phase I/II study of MK-0646, the humanized monoclonal IGF-1R
antibody in combination with gemcitabine or gemcitabine plus erlotinib
(E) for advanced pancreatic cancer. J Clin Oncol (Meeting Abstracts)
28:4039.
- Philip PA, Mooney M, Jaffe D, Eckhardt G, Moore M, Meropol N, et al.
Consensus report of the national cancer institute clinical trials planning
meeting on pancreas cancer treatment. J Clin Oncol 2009;27:5660-9.[LinkOut]
- Schilsky RL. Personalized medicine in oncology: the future is now. Nat Rev
Drug Discov 2010;9:363-6.[LinkOut]
- Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB,
et al. Frequent somatic mutations and homozygous deletions of the p16
(MTS1) gene in pancreatic adenocarcinoma. Nat Genet 1994;8:27-32.[LinkOut]
- Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum
E, et al. DPC4, a candidate tumor suppressor gene at human chromosome
18q21.1. Science 1996;271:350-3.[LinkOut]
- Redston MS, Caldas C, Seymour AB, Hruban RH, da Costa L, Yeo CJ,
et al. p53 mutations in pancreatic carcinoma and evidence of common
involvement of homocopolymer tracts in DNA microdeletions. Cancer
Res1994:54:3025-33.[LinkOut]
- Goggins M, Hruban RH, Kern SE. BRCA2 is inactivated late in the
development of pancreatic intraepithelial neoplasia: evidence and
implications. Am J Pathol 2000;156:1767-71.[LinkOut]
Cite this article as:
Huang T, Kar S, Javle M. Personalized therapy for pancreatic cancer: Myth or reality in 2010? J Gastrointest Oncol. 2010;1(1):24-33. DOI:10.3978/j.issn.2078-6891.2010.009
|





